One-track mind
/I promised myself so faithfully that I would write about neuroscience. Or at least neural-immune interactions. But it’s tricky to think about other topics when something exciting happens in vaccine research and there’s this public outlet just sitting there asking for science news… So here follows a blog post about vaccines with the crowbarred excuse that cerebral malaria is a thing that happens. Vaccines work. In terms of public health benefits, I’d put vaccination up there with antibiotics and soap. This graphic from Leon Farrant [1] earlier in the year gives us a clear idea of just how effective the vaccination programmes of the 20th Century have been. But we don’t hear much these days about new vaccines coming onto the market. One reason for this is that contemporary vaccine researchers have been left some tough nuts to crack. The viruses, bacteria and parasites still causing significant disease have evolved myriad ways to evade not only the natural immune system, but also traditional vaccination strategies. Gone are the days when scientists like Maurice Hilleman could develop multiple vaccines in a single career but, with a perseverance verging on tenacity, modern vaccine researchers are beginning to chip away at these remaining problems. Last week saw the publication of a new vaccine trial performed by Robert Seder’s group at the National Institute of Allergy and Infectious Disease, describing an important step in the development of a malaria vaccine.
Malaria is one of the toughest nuts around. This parasite has a long evolutionary history with humanity so has intimate infective knowledge of the human body. Its lifecycle is not only complicated, but involves several different stages (analogous to eggs, larvae and adults) all of which look different and live in different bodily tissues, not to mention different hosts.
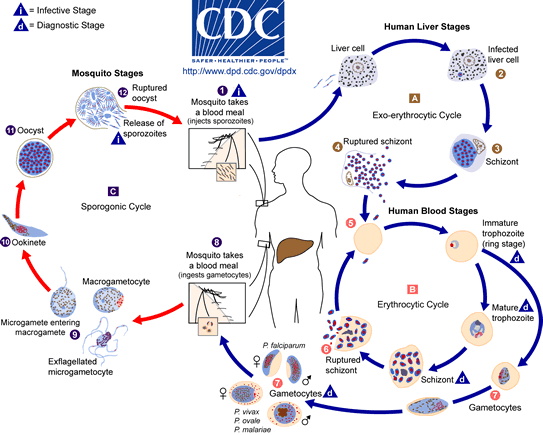
Just look at how complicated this little beast is! This kind of infection makes life very difficult for the immune system. Whether in response to an infection or a vaccine, white blood cells rely on the fact that infectious agents look very different from humans, and that the same infectious agent infecting for a second time will look the same as it did the first time. Like a master of disguise changing outfits and donning false moustaches, parasites can change their surface proteins every few days, leaving the immune system far behind in its attempts at recognition. Not only that, but they’ll squirrel themselves away inside different cells to avoid even coming into contact with white blood cells.
So how do we get ahead of this sneaky beast? Seder’s group think they have the answer [2]. Or at least one potential answer. Instead of trying to remove the parasite once it has a foothold in the body, their PfSPZ vaccine targets sporozoites – that is the parasite as it looks when it’s first injected into the bloodstream by a mosquito. PfSPZ stands for Plasmodium falciparum sporozoite. P. falciparum is one of the most common strains of malaria and is used in malaria research because (in a very rare move) the CDC allow the deliberate infection of humans with this strain in a model known as controlled human malaria infection (CHMI). Here, scientists get to test malarial vaccines in healthy humans by deliberately infecting them with malaria after vaccination and waiting to see if they get sick. To prevent full-blown disease, subjects are treated with approved anti-malarials after the test whether they get sick or not, but they usually do. Up to now, malaria vaccines have proved broadly unsuccessful. The only known method providing robust protection to date involved leaving the task of immunisation to the mosquitoes. In that study [3] conducted by the US military, scientists took mosquitoes infected with malaria, exposed them to radiation to render the parasite uninfectious and then let them bite human subjects. Over 1000 times. After 1000+ bites, over the course of up to 10 years, people were protected from subsequent CHMI. In light of the obvious difficulties and objections to this, Seder and his colleagues have now refined the original technique. It still involves mosquitoes infected with malaria, and those mosquitoes are still irradiated to knock out the parasite. But now, harmless parasites from several thousand mosquitoes can be isolated, purified and injected into people in a faster, more controlled, and less uncomfortable way. After one injection (rather than 1000 mosquito bites) the subjects are protected from subsequent CHMI. One of the things that makes this new study stand out is that the vaccine tested fully protected everyone who was given the highest dose. That is to say that the vaccine protects everyone, even when they’re deliberately injected with infectious malarial parasites.
This is exciting news for anyone in the vaccine field but still comes with some caveats. The participants in this trial were infected with malaria just 3 weeks after their last immunisation so we have no idea how long protection will last. The immunisation itself is also an issue because it only works if you inject it directly into the bloodstream (intravenously). Most vaccines are given into the muscle or skin, which is a relatively unskilled procedure. Intravenous injection requires more expertise and comes with more risk, so rolling out an intravenous vaccine to areas with poor infrastructure and limited skilled medical professionals will be tough. Having said all that, and taking into account yet more caveats given by the authors themselves, this really is exciting news. Malaria kills 0.5-1 million people every year 86% of whom are of children under 5 [4]. This vaccine may have its limitations, but it’s worth a shot.
References:
[1] http://www.behance.net/gallery/Vaccine-Infographic/2878481
[2] http://www.sciencemag.org/content/early/2013/08/07/science.1241800.full
[3] http://jid.oxfordjournals.org/content/185/8/1155.full OPEN ACCESS!
[4] http://www.who.int/malaria/publications/world_malaria_report_2012/wmr2012_no_profiles.pdf


 In this edition of Ask a Neuroscientist, we’ll answer two questions that address a similar principle: Can you train to have a better brain?
In this edition of Ask a Neuroscientist, we’ll answer two questions that address a similar principle: Can you train to have a better brain?